erfelijkheid

meer info
Voor meer info over genetische oorzaken van oogaandoeningen, klik hier.
uitleg
Linkage studies hebben de oorzaak voor het Syndroom van Wagner gelokaliseerd op chromosoom 5. Een linkage studie meet allerlei eigenschappen van het DNA van patiënten en gezonde mensen en kijkt welke eigenschap correleert met het hebben van het Syndroom van Wagner. Bijvoorbeeld: als alle mensen met Wagner bruine ogen hebben, en alle gezonde mensen niet, is de kans groot dat het gen dat voor bruine ogen zorgt, ook leidt tot het syndroom van Wagner.
Iedereen heeft in elke cel 48 chromosomen: 24 van je moeder en 24 van je vader. Wanneer deze tijdens de bevruchting samen komen, blijven ze niet netjes naast elkaar liggen, maar overlappen, breken en combineren tot 48 unieke chromosomen waarop stukjes van je vader en moeder worden afgewisseld. Totaal heb je nog wel 50% van je moeder en 50% van je vader, maar niet langer als 1 chromosoom van je vader en 1 van je moeder, maar als 2 chromosomen met duizenden kleine stukjes afwisselend van je vader en van je moeder. Aan je eigen kinderen geef je één van deze chromosomen door.
Dat maakt het mogelijk om nog veel meer in te zoomen op de plek op het chromosoom waar het gen ligt dat Wagner veroorzaakt. Wanneer Wagner nou nog steeds samengaat met het hebben van bruine ogen, betekent dat dat het Wagner gen heel dicht bij het gen voor bruine ogen ligt (of zelfs hetzelfde gen is). Als het gen voor bruine ogen namelijk ver weg van het gen dat Wagner veroorzaakt zou liggen, is de kans heel klein dat bij alle patiënten juist deze twee stukjes DNA telkens samen worden overgeërfd.
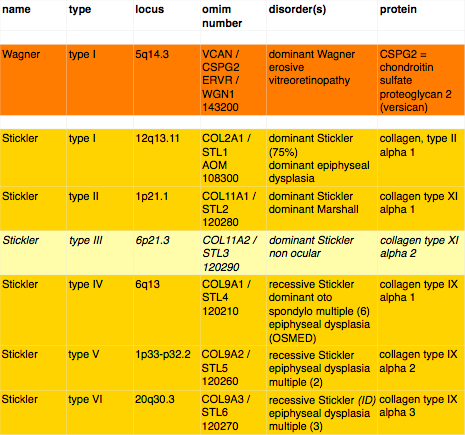
In het artikel van Graeme Black uit 1999 hebben ze de locatie voor het gen dat Wagner veroorzaakt, ook wel Wagner Disease Locus WGN1 genoemd, gereduceerd tot een klein gebied op de lange (q) arm van chromosoom 5: 5q14.3. Hierin liggen twee genen: het gen voor ‘link protein’ (CRTI.1) en het gen voor Versican (CSPG2). Verder onderzoek van familie W1 sluit het gen voor link protein uit. Het gen voor Versican lijkt een goede mogelijkheid omdat het versican eiwit belangrijk is voor de structuur van de extracellulaire matrix (het spul tussen cellen, het bindweefsel) waaruit ook het glasvocht bestaat. De logische stap is dus om te kijken of er in het Versican-gen een mutatie is bij de patienten. Nu bestaat een gen weliswaar uit één stuk DNA, maar als dat gen wordt afgelezen om er een eiwit van te maken, worden delen van het DNA overgeslagen. De delen die worden afgelezen heten exonen en de delen die worden overgeslagen heten intronen. Rondom de overgang van een intron naar een exon en andersom, zijn codes in het DNA gebouwd zodat duidelijk is welke stukken moeten worden afgelezen en welke niet. Een intron eindigt altijd met de DNA-basen/letters AG.
Het onderzoek van de Japanse groep heeft een mutatie gevonden waardoor AG is veranderd in GG. De groep van Cremers vond AG > AA. In beide gevallen is nu niet meer herkenbaar waar het intron eindigt en het exon begint, en dus zal deze ‘splice site’ worden overgeslagen. Verderop in het exon ligt ook de code AG. Normaliter wordt dit niet als splice site gezien omdat het midden in een exon ligt. Maar omdat de eigenlijke splice site wordt overgeslagen, bindt het eiwitcomplex zich nu hieraan en wordt er dus in plaats van alleen het intron ook nog het eerste stuk van het exon overgeslagen. Het eiwit dat wordt afgeschreven zal dus ook iets kleiner zijn.
Wat waarschijnlijk ook veel gebeurt, is dat ook die AG wordt overgeslagen en pas de AG site van het volgende intron wordt gevonden als einde van het intron.
page last modified Aug 9th 2014

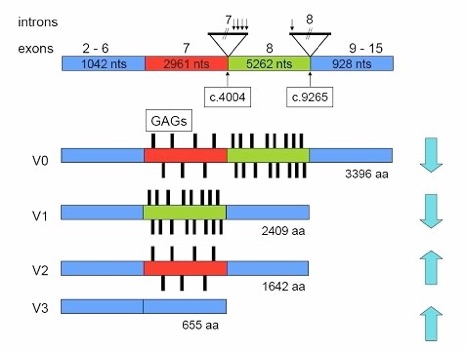
fig. Mutaties in VCAN die geassocieerd worden met vitreoretinopathy en de effecten op splice varianten en proteine isoformen.
Ook in gezonde mensen komen verschillende ‘splice-varianten’ voor. Bij het Versican eiwit komen er vier varianten voor:
V0 het hele eiwit, inclusief exon 7 en exon 8
V1 zonder exon 7
V2 zonder exon 8
V3 zonder exon 7 en exon 8
De verhoudingen van deze vier varianten is verschillend in verschillende delen van het lichaam en kan ook veranderen tijdens de ontwikkeling. In de hersenen komt bijvoorbeeld bijna alleen variant V2 tot expressie, terwijl in het hart alle varianten behalve V2 tot expressie komen. In het netvlies komen alle varianten voor, maar vooral V3 en V0. De verhoudingen in glasvocht zijn niet bekend.
De mutatie zal leiden tot een lagere expressie van variant V0, omdat exon 8 deels in zijn geheel zal worden overgeslagen. Dat leidt dus tot een hogere expressie van variant V2. Het deel van V0 dat wel tot expressie komt, zal iets kleiner zijn, omdat het begin van exon 8 is overgeslagen. Ook als variant V1 moet worden gemaakt, zal de mutatie ertoe leiden dat vaak exon 8 wordt overgeslagen. Dat leidt dan dus tot een hogere expressie van variant V3, en tot een kleiner V1 eiwit.
(met dank aan Karlijn van Aerde)
Het syndroom van Wagner is een dominant autosomale oogziekte. Dat beketent dat 50% van de nakomelingen Wagner zal hebben, jongen of meisje, dat maakt niet uit.
Het gen dat verantwoordelijk is voor het syndroom van Wagner is het gemuteerde Versican gen, liggende op de lange arm van het vijfde chromosoom 5q14.3 en wel van basepaar 82.767.529 tot 82.877.799
Wagner & Stickler: einde aan de verwarring
Voordat het gen was gevonden, was er sprake van een grote verwaring tussen Stickler en Wagner. Onderzoekers gingen er lange tijd vanuit, dat beide afwijkingen een gemeenschappelijke oorzaak hadden. Wagner werd wel aangeduid met ‘ocular-only-Stickler’. Stickler komt veel vaker voor maar werd veel later voor het eerst beschreven.
Irene Maumenee (University of Illinois, Chicago, USA) had een andere benadering. Zij ging uit van twee verschillende aandoeningen: de één (Wagner) met uitsluitend oogheelkundige aspecten, de ander (Stickler) met ook afwijkingen aan botten in het gezicht (open verhemelte) en andere afwijkingen aan het skelet.
Op dit moment zijn er 6 Stickler varianten bekend. De belangrijkste, die 75% van de Stickler-gevallen voor zijn rekening neemt, vindt zijn oorsprong in gen COL2A1 en werd voor het eerst genoemd door Clair Francomano (Baltimore, USA) in 1987, de volgende werd gevonden in 1994 door Han Brunner (Nijmegen, Nederland). Het ging om COL11A2, de enige variant zonder oogheelkundige verschijnselen. Vervolgens vond Susanna Annunen (Oulu, Finland) in 1999 de variant veroorzaakt door COL11A1. Guy Van Camp (Antwerpen, België) in 2006 vond de vierde, de enige recessieve, variant COL9A1. Ook mutaties in COL9A2 en COL9A3 bleken recentelijk te leiden tot een recessieve vorm van Stickler.
Het was duidelijk dat de Japanse en de Nederlandse Wagners iets geheel anders hadden. Het duurde tot 2005 vooraleer het Versican gen kon worden aangewezen als de oorzaak voor het syndroom van Wagner.
Erosieve vitreoretinopathie
Zoals je kunt zien wordt ook erosieve vitreoretinopathie veroorzaakt door het Versican-gen. Het wordt / werd gezien als een allele aandoening: een andere mutatie van hetzelfde gen. ERVR werd voor het eerst beschreven in 1994 door Brown et al (Department of Ophthalmology, University of Iowa, College of Medicine, Iowa City, USA). Zij noemden ERVR nog een aparte aandoening.
De Nederlandse onderzoeksgroep onder leiding van Frans Cremers vond de c.4004-5T>C variant bij 4 grote families met de ziekte van Wagner, maar ook bij de Nederlandse ERVR familie.
Genetische analyse laat zien dat al deze families dezelfde voorouder hebben met deze CSPG2/Versican variant (Mukhopadhyay et al. 2006). Dat betekent dus dat ERVR niet beschouwd kan worden als een aparte ziekte.
Daar komt nog bij dat de familie in Iowa (EFRVR) dezelfde mutatie heeft als de kleine Nederlandse familie uit de buurt van Deventer met Wagner. De Nederlandse oogarts die destijds betrokken was bij het omschrijven van de familie met ERVR zegt nu dat dit waatschijnlijk op een misverstand berust.
Er is nog geen verklaring gevonden waarom dezelfde genetische variant kan zorgen voor grote verschillen in ernst van de symptomen bij de verschillende familieleden.
Op de pagina ‘symptomen’ leggen we uit welke verschillen met Wagner zijn gemeld in families die zijn gediagnosticeerd met ERVR.
syndroom van Wagner
Het zorgt voor een afwijkende splicing naar verschillende eiwitvarianten. Het syndroom van Wagner is de enige bekende aandoening die gelokaliseerd is op het Versican gen. Een schatting leert dat deze genmutatie waarschijnlijk niet ouder is dan 1000 jaar.
Zie het plaatje en de uitleg daaronder. Links worden nog uitleg gegeven over dit soort van onderzoek.
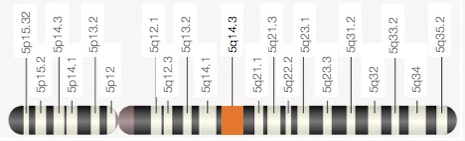
image nlm-nih / usa (modified)

wijze van overerven
Het syndroom van Wagner is een autosomaal dominante aandoening. Dat betekent dat het gen niet op het geslachts-chromosoom ligt (autosomaal) en dat één fout gen voldoende is om de ziekte te erven (dominant). In onderstaande pdf wordt de overerving uitgelegd.

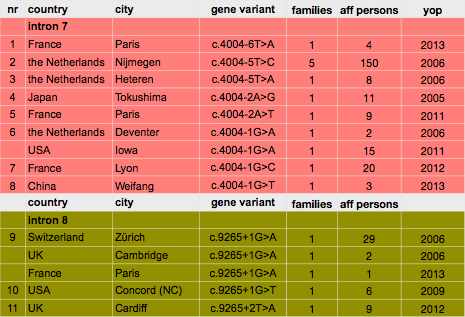
Inmiddels zijn er 11 verschillende mutaties gevonden in 14 verschillende families. In 2013 werden nog 2 nieuwe Franse families gevonden, waarvan 1 een de novo mutatie (voor het eerst gemuteerd in de gevonden persoon). In China werd in 2013 in een landelijk (!) onderzoek 1 familie gevonden met drie aangedane familieleden. Dat is opnieuw een indicatie hoe zeldzaam Wagner is.
mutaties
De mutatie bij de Nederlandse Wagners ligt in intron 7 (c.4004-5T>C) net als de mutatie bij de Japanese Wagners (c.4004-2G>A). Dus de mutatie ligt respectievelijk 5 (NL) or 2 (JP) nucleotiden vóór het begin van exon 8. Bij de Zwitserse Wagners en de Britse familie uit Cambridge ligt de mutatie in intron 8 (c.9265+1G>A). Dus de eerste nucleotide ná exon 8. Ook de nieuwe familie uit Noord-Caroline (USA) heeft een mutatie op deze ‘Hot-Spot’, maar daar is de eerste G veranderd in een T: c.9265.+1G>T.