genetische oorzaken erfelijke blindheid

page last modified Jan 2nd 2010
syndroom van Wagner
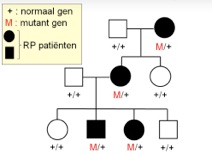
Bij een dominante ziekte draagt de patiënt een afwijking in één van de twee chromosomen waarop de mutatie zich bevindt, dus de kans dat een kind van deze patiënt dezelfde genafwijking krijgt is 50%. Bij een dominante oogziekte zien we dus meestal in meerdere generaties de oogziekte verschijnen. De term ‘dominant’ geeft aan dat de defecte versie van een eiwit (50% van het eiwitproduct) een negatief effect heeft op de normale versie (50% van het eiwitproduct) van het eiwit dat wordt gecodeerd door het gen op het ‘normale’ chromosoom.
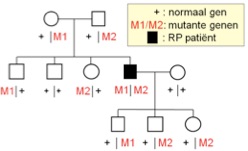
Bij een recessieve ziekte draagt de patiënt in beide genkopieën een defect. Beide ouders dragen in één van de twee genen een defect, maar ze zijn zelf gezond. De kinderen van een patiënt zijn in de regel ook gezond hoewel ze steeds één defect gen erven.
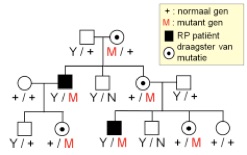
Bij X-gebonden RP zijn in de regel alleen mannen aangedaan. Zij dragen immers 1 X chromosoom, afkomstig van de moeder, en een geslachtsbepalend Y chromosoom, dat bijna geen genetische informatie bevat, afkomstig van de vader. De mannelijke RP patiënt krijgt dus zijn defect altijd van zijn moeder. De moeder is niet of mild aangedaan omdat zij ook nog een normaal X-chromosoom draagt. Een man met een X-gebonden RP zal nooit zonen krijgen met RP, hij geeft immers zijn Y-chromosoom aan zijn zonen. De dochter van een man met X-gebonden RP is draagster van het defect, maar krijgt geen RP. Een vrouwelijke draagster van een X-gebonden mutatie zal de mutatie aan de helft van de zonen doorgeven, die RP krijgen, en aan de helft van de dochters, die op hun beurt draagster zijn van het gendefect.
Duidelijkheid over erfelijkheid en herhalingsrisico in de familie
In veel situaties is de erfelijkheid duidelijk. Als in meerdere generaties van een gezin patiënten voorkomen, en zowel mannen en vrouwen zijn aangedaan, dan zal de overerving in de regel dominant zijn. Als patiënten maar in 1 generatie voorkomen, dan gaat het meestal om recessieve overerving. Het wordt lastig als maar één patiënt is aangedaan of alleen maar mannen zijn aangedaan in één generatie. Is de overerving recessief of X-gebonden of kan het allebei? Stel er is nog kinderwens. Wat is de kans voor een volgend kind om RP te krijgen?
Het kan gaan om recessieve overerving: beide ouders dragen één mutatie; de kans dat een volgend kind is aangedaan is dan 25%. Het kan ook een X-gebonden defect zijn: de moeder is drager van een recessief defect maar heeft ook een gezond gen en heeft dan geen RP. Bij een volgend mannelijk kind is de kans 50% dat deze RP krijgt; bij een vrouwelijk kind is de kans 0% dat zij RP krijgt, maar 50% dat zij een gezonde draagster is van de mutatie. Soms kan het ook om dominante overerving gaan omdat er van de patiënt geen familiegegevens bekend zijn (bv. adoptie). In dat laatste geval is de kans dat een kind van de patiënt RP krijgt 50%.
Conclusie: Om een onderscheid te kunnen maken tussen deze vormen van overerving en herhalingsrisico’s moeten we dus het gendefect eerst opsporen.
Betere voorspelling ziektebeloop
Kan het vinden van een mutatie de oogarts helpen om te voorspellen hoe de ziekte zich zal uiten in de toekomst? Ik geef hierbij een voorbeeld van een familie die door collega Arthur Bergen (NIN, Amsterdam) is bestudeerd. We hebben wederom te maken met een jong-volwassen mannelijke patiënt waarbij aanvankelijk geen familieleden met blindheid bekend waren. Hij kon minder goed zien, had moeite met kleuren zien en een test toonde aan dat kegeltjes, betrokken bij het kleurenzien, niet goed functioneerden. Na enig speurwerk bleek dat een broer van zijn grootmoeder en een paar achterneven ernstige RP hadden. Dit wijst er zeer sterk op dat het gaat om X-gebonden RP. De oudoom bleek op oudere leeftijd nog maar 1% gezichtsveld te hebben. Naast de kegeltjes, waren ook de staafjes aangedaan. Bij X-gebonden RP is de prognose slecht omdat de ziekte tussen het 30e en 40e levensjaar snel voortschrijdt. Hoewel het geen goed nieuws was voor de jongeman, kon wel worden aangegeven hoe de ziekte zich zal ontwikkelen. Niet iedereen zit mogelijk op een dergelijk bericht te wachten, maar in principe is het mogelijk om betere informatie te geven waardoor soms betere studie- of werkkeuzen gemaakt kunnen worden.
Beter advies over voedingssupplementen
Heeft kennis van het gendefect een invloed op het gebruik van voedingssupplementen? Reeds vele jaren geleden heeft dr. Eliot Berson in Boston een grote studie gedaan waarin hij heeft gevonden dat vitamine A een klein gunstig effect heeft bij een heterogene groep RP patiënten. Hij wist destijds niet welke gendefecten er speelden.
Om licht om te zetten in een signaal naar de hersenen is het stofje rhodopsine erg belangrijk. Rhodopsine bestaat uit twee delen; een eiwit deel genaamd opsine en een molecuul dat veel lijkt op vitamine A, en daar ook van is afgeleid, genaamd retinal. Samen vormen ze rhodopsine. Retinal kan alleen via vitamine A of aanverwante moleculen die we via voeding binnen krijgen, worden gemaakt. Als een lichtfoton rhodopsine raakt, verandert de retinal component en het gehele eiwit van vorm en wordt een signaal gegeven aan de hersenen. Dit is de basis van ons zien. Bij patiënten met een verandering (mutatie) in het rhodopsine gen, kan aanvullende vitamine A inname een overweging zijn.
Bij andere ziektebeelden kan een teveel aan vitamine A schadelijk zijn zoals bij RP veroorzaakt door het ABCA4 gen. Door mutaties in het ABCA4 gen hoopt inactief retinal zich op in het netvlies en sterven de netvliescellen langzaam maar zeker. Dit betekent dat teveel retinal, en dus ook teveel vitamine A, dat als grondstof fungeert voor het maken van retinal, schadelijk is voor het netvlies. Kortom, teveel retinal is niet goed voor de netvliescel; als er echter te weinig is zal er geen signaal worden doorgegeven aan de hersenen.
Met andere woorden, alleen indien we het gendefect weten kunnen we voorspellen of extra vitamine A gunstig of ongunstig zou kunnen zijn.
Duidelijkheid over genetische oorzaak in familie
Door onvoldoende kennis over de erfelijkheid is er bij ouders van patiënten vaak sprake van een ‘schuldbesef’ t.a.v. de oorzaak van de oogziekte bij hun kind, met name in geval van recessieve overerving. Soms worden de oorzaken gezocht in niet-genetische factoren. Ieder mens draagt diverse recessieve gendefecten die niet tot uiting komen omdat er in de meeste gevallen nog een ‘normale’ kopie van het gen aanwezig is. Het is toeval dat de ouders van een kind beiden defecten dragen in hetzelfde gen en deze allebei doorgeven aan hun kind waardoor die bv. RP krijgt. Uitleg over de erfelijkheid door een klinisch geneticus aan de hand van de gevonden erfelijke defecten kan onnodig schuldbesef wegnemen.
Opsporing kandidaten voor genetische therapieën
Een andere reden om de gendefecten te weten heeft te maken met de ontwikkelingen op het gebied van gentherapie. RPE65 gentherapie staat volop in de schijnwerpers, maar mutaties in RPE65 verklaren niet meer dan 1% van de erfelijke blindheid in Nederland. Hoe zijn de vooruitzichten van gentherapie voor andere RP genen?
Een belangrijke tussenstap van genidentificatie naar therapie bij de mens is het maken van een diermodel, in de regel een muismodel. Voor 18 van de 27 bekende recessieve en X-gebonden RP genen zijn diermodellen gemaakt en in 6 hiervan is bij de muis succesvol gentherapie toegepast. Veel onderzoekers hebben zich laten inspireren door de resultaten bij patiënten met RPE65 mutaties en wij verwachten dat de toepassingen bij de mens de komende jaren langzaam maar zeker toenemen. Voor gentherapie zoals in het geval van het RPE65 gen moet men dus exact weten in welk gen het defect ligt.
Er zijn nog andere therapieën in ontwikkeling waarbij het niet belangrijk is welk gen de mutatie draagt, maar wel welk type mutatie het betreft. Eiwitten worden in 2 stappen gemaakt. Van het DNA (de chromosomen) wordt een kopie gemaakt, genaamd RNA. Het RNA dient als code voor het maken van een eiwit. Een eiwit kunt u zich voorstellen als een kralenketting waarbij er 20 verschillende kleuren kralen zijn. Onderzoekers noemen deze kralen aminozuren. Het RNA bevat aan de voorzijde een ‘start’ signaal en aan de achterzijde een ‘stop’ signaal. Hierdoor heeft de kralenketting een bepaalde lengte. Bij sommige erfelijke ziekten bevindt zich in het DNA, en daarmee ook in het RNA, een extra ‘stop’ signaal. In een netvliescel, bv. een staafje, zal dan maar een deel van het eiwit worden gemaakt, waardoor het eiwit geen normale functie meer heeft. Er bestaat een stofje, genaamd PTC124, dat er voor zorgt dat het stopsignaal als het ware wordt genegeerd.
Er komt een willekeurige kraal op de plaats van de mutatie en het eiwit kan wel volledig worden gemaakt.
Het PTC124 is al getest bij een diermodel van een erfelijke spierziekte en bij gezonde vrijwilligers. Het lijkt weinig of geen bijwerkingen te hebben. PTC124 is geen wondermiddel. Het verliest in 6 uur de helft van zijn activiteit en moet dus regelmatig worden toegediend. Het effect van PTC124 is beperkt. Indien een eiwit door twee recessieve mutaties volledig geïnactiveerd wordt, dan is de restactiviteit van het gemuteerde gen/eiwit nagenoeg 0%. Men hoopt dit met PTC124 op te krikken naar 10%. Dat lijkt weinig maar zou voor veel netvlieseiwitten voldoende kunnen zijn om het ziekteproces te vertragen of te stoppen.
PTC124 werkt dus alleen als er sprake is van een bepaald type mutatie, nl. een zogenaamde stopmutatie. Ongeveer 1/3 van alle patiënten met erfelijke aandoeningen draagt 1 of 2 mutaties van dit type, dus in potentie zijn de toepassingen voor alle erfelijke ziekten zeer groot. Het is moeilijk een voorspelling te doen hoe snel en in welke vorm PTC124, of een hierop lijkend molecuul, als ‘geneesmiddel’ beschikbaar komt. Mocht het klinisch toegepast gaan worden, dan is het voor iedere patiënt met een recessieve ziekte van belang om te weten of men al dan niet stopmutaties draagt.
Opsporing erfelijke oorzaken RP en verwante ziektebeelden
Hoeveel is er eigenlijk bekend over de oorzaken van erfelijke blindheid in Nederland? Wij schatten dat we in theorie 50% van alle oorzakelijke genen hebben gevonden. Als voorbeeld gebruiken we hierbij RP, de meest voorkomende erfelijke oogziekte. Wij schatten dat er in Nederland 4000 RP patiënten zijn (d.w.z. dominante, recessieve, en X-gebonden RP samen). We kennen 47 oorzakelijke genen die samen 50% van de ziekte verklaren. Wij denken dat er in Nederland bij maximaal 400 patiënten de daadwerkelijke oorzaak bekend is, dus minder dan 10%. Er gaapt dus een groot gat tussen theoretische kennis en het weten van de oorzaak bij de patiënten. Waarom is dat zo? Onderzoekers worden afgerekend op het vinden van nieuwe genen; er is geen of weinig geld voor het vinden van mutaties in bekende genen. In DNA diagnostiek centra in Amsterdam en Nijmegen wordt wel getracht systematisch te zoeken naar de genetische oorzaken. De vraag naar deze diagnostiek is nog beperkt omdat er in de meeste families geen erfelijkheidsvraagstellingen zijn, deze testen tot voor kort te duur waren, en er geen therapieën bestonden. Daar komt nu langzaam verandering in. Hoe gaan we dit de komende 5 jaar aanpakken? Wij willen ons dus concentreren op RP, waarvan het grootste deel van de patiënten een recessieve overerving heeft (schatting: 3000 patiënten in Nederland). Zoals gezegd kennen we in theorie de helft van de genetische oorzaken. Wij stellen voor dat RP patiënten tijdens hun reguliere bezoek aan de oogarts worden voorgelicht over ons diagnostisch onderzoek en gevraagd worden bloed af te staan. In de diagnostische centra kan men testen op het aanwezig zijn van mutaties in de bekende genen, en als deze niet worden gevonden, dan zullen wij verder gaan met onderzoek om nieuwe oorzaken op te sporen.
Wij willen hierbij alle academische en een aantal perifere oogklinieken betrekken. In de afgelopen 10 jaar hebben wij in Nijmegen en Rotterdam reeds DNA verzameld van 250 recessieve RP families. Wij zullen de komende 2 jaar 750 extra recessieve of sporadisch voorkomende RP patiënten en hun familieleden betrekken bij deze studie. Daarnaast kunnen ook patiënten met de aan RP verwante oogziekten, zoals aangeboren blindheid, kegelstaaf dystrofie, en kegel dystrofie, zich aanmelden voor dit onderzoek. Alle patiënten worden eerst getest op het voorkomen van mutaties in de bekende blindheidgenen. De ontwikkelingen op dit gebied gaan zo snel dat wij verwachten dat we binnen 6 maanden bij 50% van de patiënten de oorzakelijke mutaties kunnen aantonen. Indien er geen mutaties worden gevonden in de bekende blindheidgenen, dan hanteren wij in de onderzoeksgroep de nieuwste methoden om nieuwe oorzaken op het spoor te komen. Met het vinden van nieuwe oorzakelijke genen voor erfelijke blindheid zal een allesomvattende analyse van de bekende blindheidgenen in de toekomst steeds effectiever worden.
Samenstelling en voordrachten
Prof. dr. Frans P.M. Cremers, hoogleraar Ophthalmogenetica,
afdeling Antropogenetica, UMC St Radboud, Nijmegen
Drs. Karin W. Littink, promovendus, afdeling Antropogenetica,
UMC St Radboud, Nijmegen en Oogziekenhuis, Rotterdam
Dr. Anneke I. den Hollander, universitair hoofddocent Multifactoriële Oogziekten, afdeling Oogheelkunde, UMC St Radboud, Nijmegen
Dr. L. Ingeborgh van den Born, oogarts, Oogziekenhuis, Rotterdam.
dominante overerving
recessieve overerving
x-gebonden overerving
In het najaar van 2009 werden door een viertal wetenschappers voordrachten gehouden voor patiënten en familie, georganiseerd door NVBS (Retinabelangen) en Retina Nederland. Hieronder tref je een samenvatting aan.
In de afgelopen 22 jaar heeft de ophthalmogenetica onderzoeksgroep in Nijmegen zich succesvol beziggehouden met het opsporen van nieuwe genen voor erfelijke oogziekten. Samen met de inspanningen van vele collega’s wereldwijd heeft dit geresulteerd in meer dan 150 oorzakelijke genen op dit moment, die, afhankelijk van het ziektebeeld, tussen de 40 en 90% van de oorzakelijke defecten omvatten. Dat betekent niet dat wij de daadwerkelijke oorzaken bij zoveel individuele patiënten in Nederland hebben opgespoord. Wij schatten dat minder dan 10% van de Nederlandse patiënten met erfelijke oogziekten weten welk gendefect(en) zij dragen. Wij stellen het ambitieuze doel voor om in 2025 het overgrote deel van de oorzaken van erfelijke blindheid in Nederland in kaart te hebben gebracht. Als eerste stap willen wij in de komende 5 jaar proberen om in totaal 1000 RP patiënten de genetische oorzaak op te sporen. Wat is het nut hiervan en waarom is er de noodzaak om dit te willen doen?
Allereerst vanuit het perspectief van de onderzoekers:
-
1.Indien we nieuwe kennis vergaren over oogziekten komen we vanzelf meer te weten over het normale functioneren van het oog. De fundamentele kennis neemt daardoor toe.
-
2.Omdat er zoveel verschillende genetische oorzaken zijn, zal het ook niet gemakkelijk zijn om voldoende aantallen patiënten op te sporen voor bv. gentherapie studies. Het systematisch opsporen van de oorzaken van erfelijke blindheid resulteert in grote patiënt-groepen die ook klinisch goed in kaart kunnen worden gebracht en met elkaar vergeleken kunnen worden.
Vanuit het perspectief van de patiënt en diens familie zijn er tenminste 5 redenen waarom de systematische opsporing van gendefecten belangrijk is:
-
1.In een deel van de families levert het kennen van de mutaties nieuwe informatie op over de erfelijkheid.
-
2.Soms zal het vinden van een mutatie het ziektebeloop beter kunnen voorspellen.
-
3.In een subgroep van patiënten zal de arts een gewogen advies kunnen geven omtrent bijvoorbeeld voeding.
-
4.In veel gevallen zullen de ouders van een patiënt met een recessieve oogziekte graag willen weten hoe de erfelijkheid in hun familie er uit ziet en is het soms belangrijk om de diagnose van een ziekte die in de rest van de familie niet voorkomt te bevestigen.
-
5.Het kennen van de mutatie kan op termijn belangrijk zijn voor het toepassen van nieuwe therapieën.
Basisprincipes erfelijkheid
Om punt 1. te illustreren volgen eerst de basisregels van de erfelijkheid. In een bevruchte eicel bevinden zich 23 chromosomen van vader en 23 chromosomen van moeder. In iedere lichaamscel, of dat nu een huidcel is of een netvliescel, bevinden zich dus 23 paar = 46 chromosomen. De chromosomen zijn genummerd van 1 tot 22, van groot naar klein. Daarnaast zijn er nog het X-chromosoom en het Y-chromosoom. Het X-chromosoom komt 2x voor bij de vrouw; de man draagt 1 X en 1 Y-chromosoom. Op al deze chromosomen samen bevinden zich circa 25.000 genen. Ieder gen bevat de code van een apart eiwit. Eiwitten zijn de bouwstenen van ons lichaam. We hebben dus van alle chromosomen, behalve het X-chromosoom bij de man, twee kopieën. We hebben dus ook van ieder gen twee kopieën.

Met dank aan
Frans Cremers
Karin Littink
Anneke den Hollander
Ingeborgh vd Born
Wat is de relevantie voor mensen met Wagner? Allereerst de uitleg over het verschil tussen dominante, recessieve en x-gebonden overerving.
Wagner is autosomaal (niet op een geslachts-chromosoom) en dominant.
Verder zie je in de literatuur dat bij sommige patiënten met Wagner een pseudo-retinitis pigmentosa wordt geconstateerd. Dat betekent dat sommige oogartsen of onderzoekers verschijnselen zien bij mensen met Wagner die lijken op RP. Dat houdt dan meestal verband met beperkt gezichtsveld en moeite met zien in het donker (nyctalopia)
En tenslotte: gen- en andere nieuwe therapieën voor erfelijke oogaandoeningen, kunnen ook voor mensen met Wagner belangrijk zijn.



Het syndroom van Wagner is een autosomale dominante aandoening. Autosomaal betekent dat de genmutatie niet op het geslachts-chromosoom ligt.