molecular research
As far as we know molecular research is being done in Zürich, Nijmegen and Tokushima.
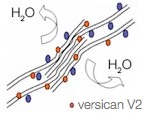
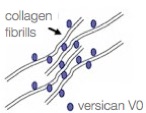
In Zürich (Switzerland) they are in the process of trying to explain the pathology of the splice mutation. They are testing hypotheses of either haplo-insufficiency or disturbed isoform ratios by analyzing the expression of various VCAN transcript and protein isoforms. In the healthy vitreous, glycosaminoglycan-rich versican isoforms prevent clumping of collagen fibrils. Splicing defects in Wagner disease lead to drastic over expression of variant V2 carrying reduced attachment site for glycosamino glycans. This situation may lead to early liquefaction of the vitreous. See the images in the left column.
They are also analyzing a -Dutch- Wagner vitreous biopt. They want to make a quantitative statement about the expression of versican at the protein level by trying to isolate proteins from the virteous biopt. They are able to identify some of the naturally occurring variants of versican, due to variant specific antibodies that the Zimmermann group in Zurich has isolated. If protein levels mimick transcript levels, they expect variant 2- based on the transcript analysis published by Frans Cremers- to be overexpressed.
And finally they are in the process of finding a mouse model for Wagner disease.
On their website they offer genetic testing.
In Nijmegen (the Netherlands) researchers are looking for intronic mutations in the Dutch family from Chinese origin from the article from Arijit Mukhopadhyay. They hope for an explanation in severity in symptoms between different family members and between members of different pedigrees with Wagner syndrome.
They too offer genetic testing.
In Tokushima (Japan) the researchers speculate that the c.4004-2A>G mutation in the CSPG2/versican gene may result in insufficient interactions between versican and various vitreous components, including HA (hyaluron acid) and type II collagen and thereby produce premature syneresis and degeneration in the vitreous gel. To test this hypothesis, functional experiments using mutant and wild-type versican cDNAs are under way in their laboratory.
phenotype - genotype
In Nijmegen they just started to more accurately phenotype Wagner syndrome to see if a connection can be made between the different mutations and difference in expression. They hope to find a predictive factor for severity in symptoms and adequate, evidence based, (prophylactic) treatment.
miscellaneous
In Chicago Irene Maumenee is doing research on connective tissue disorders and glaucoma. She did research on the original Swiss pedigree in the 19-eighties.
In India Arijit Mukhopadhyay (from the article in mol. vis. 2006 on the Versican gene in the Dutch pedigree) is doing research on the role of the Versican gene in the trabecular meshwork and ciliary body.